Workshop 2. High Throughput Sequencing Application Session¶
Overview¶
In this workshop we will accomplish the following tasks:
- Download sample FASTQ files from figshare using
wget - Downsample a FASTQ file for use in code prototyping
- Parse FASTQ file and identify good reads
- Deduplicate good reads
- Trim deduplicated reads to remove random sequences and
CACA - Calculate unique sequence counts after deduplication and trimming
- Record read count statistics of total reads, good reads, deduplicated reads, and unique reads
- Make your python script generic and apply your code to the full datasets
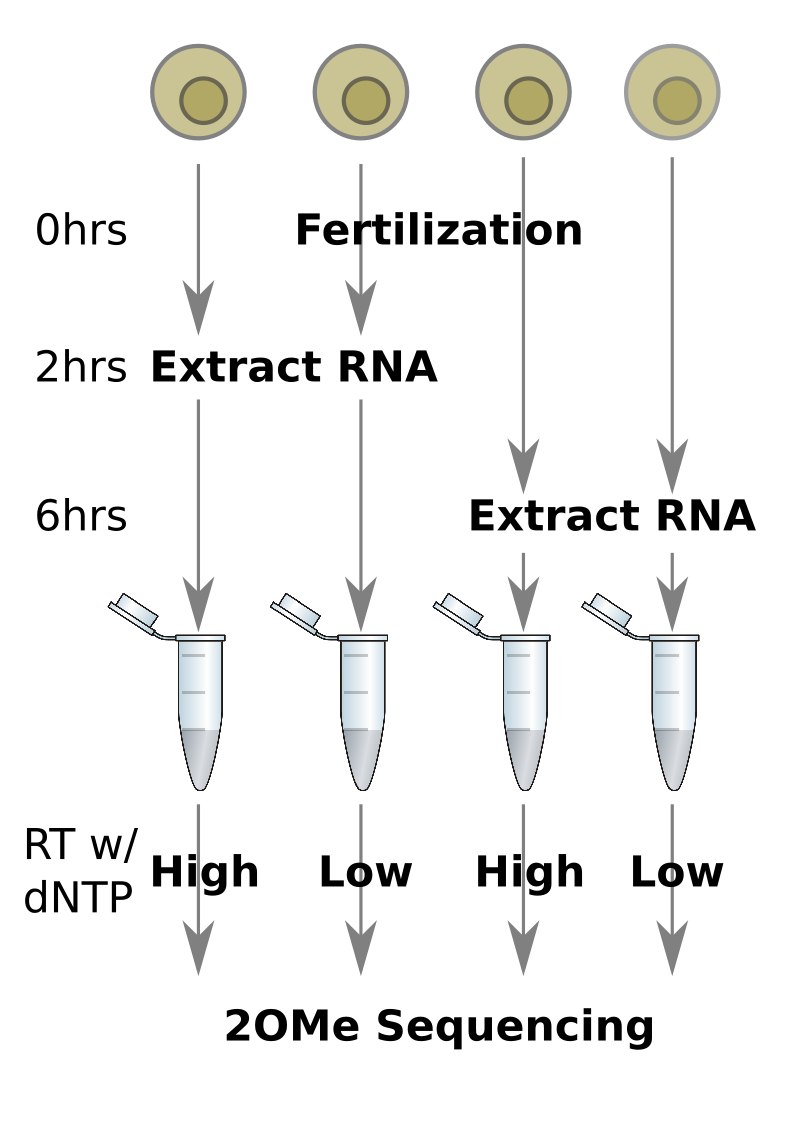
The experimental setup that produced this data is as follows:
Download sample FASTQ¶
The FASTQ files for this workshop are hosted on figshare. figshare is a free, open web platform that enables researchers to host and share all of their research files, including datasets, manuscripts, figures, posters, videos, etc. There are four FASTQ datasets hosted on figshare that we will use in this workshop:
| Sample name | Hours after fertilization | RT dNTP concentration |
|---|---|---|
| FO-0517-2AL | 2 | low |
| FO-0517-2AH | 2 | high |
| FO-0517-6AL | 6 | low |
| FO-0517-6AH | 6 | high |
The data are available here, but don’t go all clicky downloady yet.
While we could click on the link above and download the data using a web
browser, it is often convenient to download data directly using the CLI. A tool
we can use to do this is wget, which stands for ‘web get’. To download a
file available by URL, you can run the following:
$ wget https://url/to/file
This will download the file at the given URL to the local directory, creating a
local filename based on the last part of the url (e.g. file in this example).
The link to download this dataset is:
https://ndownloader.figshare.com/articles/5231221/versions/1
Tip
If you don’t have wget, you could also try curl
Running wget with this link will create a file named
1, which isn’t very nice or descriptive. Fortunately,
wget has a command line option -O <filename> that we can use to rename the
file it downloads.
Task - Download source data
Use wget and the appropriate command line arguments to download the
dataset to a file named 2OMeSeq_datasets.zip. Once the file has been
downloaded (it should be ~1Gb in size), unzip it with the unzip command.
Downsample a FASTQ file¶
If the previous task completed successfully, you should now see four files in your current working directory:
$ ls *.fastq.gz
DC-0517-2AH.10M.fastq.gz DC-0517-2AL.10M.fastq.gz
DC-0517-6AH.10M.fastq.gz DC-0517-6AL.10M.fastq.gz
Use ls to examine the size of these files, and note that they are somewhat
large.
When writing analysis code for large sequencing datasets, it is often beneficial
to use a smaller, downsampled file to enable more rapid feedback during
development. We can easily produce a smaller version of these FASTQ files using
head, I/O redirection >, and zcat, a command we haven’t covered yet.
gzip and zcat¶
Raw FASTQ files are usually very large, so, rather than store them as regular
text files, these files are often compressed into a binary format using the
gzip program. gzipped files often end in .gz, which is the case for our
sample files. Since the compression algorithm produces a binary file, we cannot
simply print a gzipped file to the screen and be able to read the contents like
we would with a text file, e.g. using cat. If we do wish to view the
contents of a gzipped file, we can use the zcat command, which merely
decompresses the file before printing it to the screen.
Warning
FASTQ files often have hundreds of millions of lines in them. Attempting to
zcat an entire FASTQ file to the screen will take a very long time! So,
you probably don’t want to do that. You might consider piping the output to
less, however.
Tip
If you’re having trouble with zcat, you could also try gunzip -c
Task - Create a downsampled file with 100k reads
Recalling the FASTQ format has four lines per read, use zcat, head,
the pipe |, and I/O redirection > to select just the top 100k reads
of one source FASTQ file and write them to a new file. You may choose any
one of the source files you wish, just be sure to give the file a unique
name, e.g. FO-0517-6AL_100k.fastq. Once you have done this, compress the
file using the gzip command, e.g. gzip FO-0517-6AL_100k.fastq.
Parse FASTQ file and identify good reads¶
Using the downsampled FASTQ file from above, we are first going to examine the
reads to determine which are ‘good’, i.e. end with the sequence CACA.
Task - Identify good reads
Write a python script, e.g. named process_reads.py, that opens the
downsampled gzipped FASTQ file, iterates through the reads, count the number
of total reads in the dataset, counts reads that end in the sequence
CACA, and retain the sequences for those reads.
HINT: python can open gzipped files directly using the gzip.open function.
Deduplicate good reads¶
In your script you should now have a set of sequences that correspond to good reads. Recall the adapter strategy caused these reads to be as follows:
4 random nucleotides
|
| True RNA fragment insert
| |
| | 2 random nucleotides
| | |
| | | Literal CACA
| | | |
v v v v
NNNNXXXXXXXXXXXXXXXXXXXXXXXXXNNCACA
In principle, this strategy should enable us to distinguish reads that are the result of PCR amplification from independent events. Specifically, reads with exactly the same sequence are very likely to have been amplified from a single methylation event (why is this?). Therefore, we are interested in eliminating all but one of reads that have exactly the same sequence.
Task: Deduplicate reads
Collapse the good reads you identified such that there is only one sequence per unique sequence. HINT: look at the set python datatype.
Trim deduplicated reads¶
The deduplicated reads represent all of the unique RNA fragments from the original sample, but they still contain nucleotides that were introduced as a result of the preparation protocol. We will now trim off the introduced bases and write the result to a FASTA formatted file.
Task - Trim off artificial bases and write to FASTA format
Using the deduplicated reads identified in step 4, trim off the bases that were introduced by the protocol. Write the resulting sequences to a FASTA formatted file.
Take the sequence of some of these reads and search BLAST for them. What species did these sequences come from?
Calculate unique sequence counts¶
Now that we have the unique RNA sequences identified by the experiment, one question we can ask is: which sequences do we see most frequently? To do this, we can count the number of times we see each identical sequence, and then rank the results descending. We can write out a list of sequences and the corresponding counts to file for downstream analysis.
Task - Count the occurence of the deduplicated sequences
Loop through the deduplicated sequences and keep track of how many times each unique sequence appears. Write the output to a tab delimited file, where the first column is the unique sequence and the second column is the number of times that sequence is seen.
HINT: Look at the csv module in the standard python library.
HINT: Look at the Counter class in the collections module of the standard python library.
Record read count statistics¶
Looking at the downsampled data, we can consider four quantities:
- # of total reads (100k)
- # of good reads
- # of deduplicated reads
- # of unique deduplicated reads
Comparing these numbers may give us an idea of how well the overall experiment worked. For example, the fraction of good reads out of the total reads gives us an idea of how efficient the sample preparation protocol is. The fraction of deduplicated reads out of the good reads tells us how much PCR amplification bias we introduced into the data.
Task - Record read count statistics
In whichever format you desire, write out the counts of total reads, good reads, deduplicated reads, and unique deduplicated reads to a file.
Make your python script generic¶
Now that we have prototyped our script using a downsampled version of the data, we can be more confident that it will work on a larger dataset. To do this, we can make a simple modification to our script such that the filename we are using is not written directly into the file, but rather passed as a command line argument. The argv property in the standard python sys module makes the command line arguments passed to the python command available to a script.
Task - Make your script generic
Use the sys.argv variable to enable the script to accept a fastq filename as the first argument to the python script. Make sure when your script writes out new files, the filenames reflect the filename passed as an argument.
Run your script on all four of the original FASTQ files and compare the results. These files are substantially larger than the downsampled file, so this may take some minutes.
Questions and Wrap-up¶
In this workshop, we have taken raw sequencing data from a real-world application and applied computational skills to manipulate and quantify the reads so we can begin interpreting them. The FASTA sequences of deduplicated reads may now be passed on to downstream analysis like mapping to a genome and quantification. The unique sequence counts we used may be also used to identify overrepresented sequences between our different conditions. When you have finished running your script on all of the full FASTQ datasets, compare and interpret the results.