Workshop 5. NGS Application Session 2¶
In this workshop, we will resume analysis of the 2OMeSeq datasets we worked with in Workshop 2. We will begin with the deduplicated FASTA sequences for the 10M sequence datasets for all four time points, which will be made available to you.
The tasks we will perform in this workshop are:
- Create an index for the zebrafish genomic ribosomal RNA sequences and align
the deduplicated FASTA sequences to it using the alignment program
bwa. - Convert the alignments from SAM format to sorted BAM format using
samtools - Count the number of alignments across all positions in the rRNA sequences
using
bedtools genomecov - Plot read count distributions using a tool of your choosing
- Compute an enrichment score for all positions in the rRNA to identify differences between low and high dNTP concentration conditions
Genomics tools¶
An introduction to the three tools we will be using in this workshop:
bwa- Burrows Wheeler Transform reference based sequence alignmentsamtools- perform operations on SAM and BA formatted alignment filesbedtools- suite of tools for doing ‘genomic arithmetic’
My video capture program was crashing like it was its job, so these materials are text-based.
bwa¶
bwa is a short read alignment program that uses the Burrows-Wheeler Transform (BWT) to encode long sequences, like a genome, into a form that is easy to search.
bwa requires an index to run, which is a set of files that contain a set of
BWT encoded sequences that the program can understand. To create a bwa index,
all we need is a FASTA formatted file of reference sequences. For example, if
we have the FASTA file zebrRNA.fa, we can create an index using:
bwa index zebrRNA.fa
This will create a new set of files with the zebrRNA.fa prefix and various
suffixes added:
$ ls zebrRNA.fa*
zebrRNA.fa zebrRNA.fa.amb zebrRNA.fa.ann zebrRNA.fa.bwt zebrRNA.fa.pac zebrRNA.fa.sa
The newly created files (e.g. zebraRNA.fa.amb) are not specified directly to
bwa, but rather specified as a index base path corresponding to the common
prefix of those files, which in this case is zebrRNA.fa.
The recommended command for aligning sequences against a preexisting index is
bwa mem (there are other modes, but mem is usually the best). The bwa mem
command is invoked with two arguments, the index base and a (possibly gzipped)
FASTQ or FASTA file. For example, if we have short reads in the sample.fasta.gz
file, we could align them against the index we built with:
bwa mem zebrRNA.fa sample.fasta.gz > sample.sam
By default, bwa mem prints out alignments to stdout, so the alignments are
redirected to a new file named sample.sam. This new file contains read
alignment information in SAM format, which is the current standard file format
for storing alignments.
samtools¶
samtools is a suite of programs that is used to manipulate SAM files. The
SAM format is a text-based format, and can become very large for short read
datasets with millions of reads. Therefore, a binary version of SAM files called
BAM (Binary SAM) files can be created using samtools view.
samtools view sample.sam -b > sample.bam
The -b argument tells samtools to output the alignments in sample.sam to
BAM format. Like bwa, samtools outputs to the stdout by default, so a
redirect is used to capture this output to a file.
Warning
Viewing a binary file to stdout without capturing it, such that it is printed to the screen, is very scary. But not dangerous.
The alignments in sample.bam are in the same order as those in sample.sam
which are roughly in the same order as they appear in sample.fasta.gz. For
some operations, it is useful or necessary to have the alignments sorted by
genomic position. We can use samtools sort to do this:
samtools sort sample.bam -o sample_sorted.bam
The alignments in sample_sorted.bam will be ordered by ascending genomic
alignment coordinate.
Using pipes, all of these operations can be done in a single command. This avoids writing large intermediate files, like SAM files, to disk. We could do all of the previous commands as follows:
bwa mem zebrRNA.fa sample.fasta.gz | samtools view -b | samtools sort -o sample_sorted.bam
bedtools¶
bedtools is another freely available program that allows analysts to perform
so-called genomic arithmetic. This essentially means dividing up the genome into
different parts and applying operations on the reads that map to them. bedtools
has many different subcommands, but the one we will use in this workshop is called
genomecov. This subcommand accepts a set of alignments and returns a histogram
of reads that map to each position in the genome.
To use genomecov, we need an input set of alignments in BAM format and a file
that contains the reference sequences and their lengths (i.e. number of nucleotides)
called a sizes file. The sizes file expected by bedtools is a tab delimited
file with two columns, the first being the name of the sequence and the second
the length of the sequence in nucleotides (see the UCSC hg19 file as an example).
If we create such a file named zebrRNA.fa.sizes using the zebrRNA.fa
file we used as the basis for our bwa alignments, we can count the number of
reads that map to each location in the ribosomal RNA index as follows:
bedtools genomecov -d -ibam sample_sorted.bam -g zebrRNA.fa.sizes > sample_sorted_coverage.tsv
The file sample_sorted_coverage.tsv will now contain the number of reads that
map to each position in our reference in a tab delimited file. These counts can
then be easily read by scripts for further analysis.
Technically speaking, the above command reports the number of sequenced bases
that map to each position of the reference. In other words, if you sum up the
counts returned, the summed value will approximately equal the number of aligned
reads times the read length. If you are interested instead to know the number
of distinct alignments that align to the reference, you can provide the -5
(or -3) option to bedtools genomecov. This will report the number of
alignments that begin (or end) at each base position. We will use -5 in the
workshop.
Workshop Problem Statement¶
An introduction to two new concepts we need to understand the goal of the analysis tasks of the workshop:
- 5’ read coverage - counting just the 5’ locations of aligned reads
- enrichment score - an algorithm for quantifying the overabundance of 5’ read alignments compared to surrounding bases for every position in the reference
Recall that in the 2OMeSeq protocol, reverse transcription stalls immediately upstream of methylation events under low dNTP conditions, resulting in an enrichment of reads that begin at those locations. Under normal dNTP conditions, no such enrichment should occur. We can use this principle to identify putative methylation events programmatically by analyzing the 5’ read alignment patterns across the transcriptome. The following figure illustrates this process:
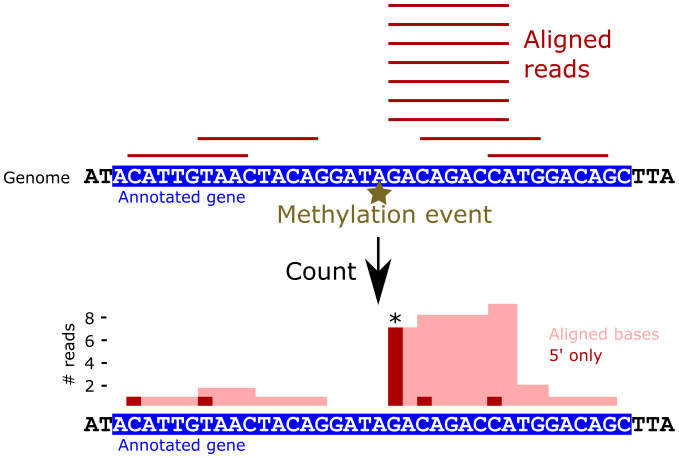
In the figure, by counting just the 5’ ends of the alignments (dark red bars), the methylation event is much more clearly marked than when considering all aligned bases (light red area).
We can use the 5’ end alignment counts to calculate a score at each base position that identifies locations that show high read pileup within a surrounding window as illustrated in the next figure.

The intuition behind the score is that individual positions with many more 5’ alignments than the average number of reads per base within a window surrounding that position are putative methylation events.
In this workshop, you analyze the deduplicated reads we identified in workshop 2 and identify putative methylation sites in the zebrafish ribosomal RNA genes.